
Second-Principles Density Functional Theory
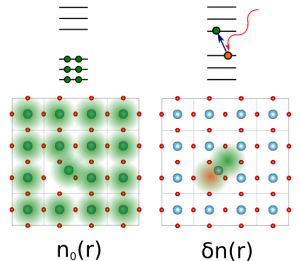
The SPDFT approach is a large-scale electronic structure method, prepared to deal with 100000+ atoms, based on a systematical approximation of the full DFT energy and with a similar level of accuracy. This is achieved by separating the total density into a reference part, typically corresponding to the system’s neutral, geometry-dependent ground state, and a deformation contribution that takes into account the difference between the full density and the reference one.
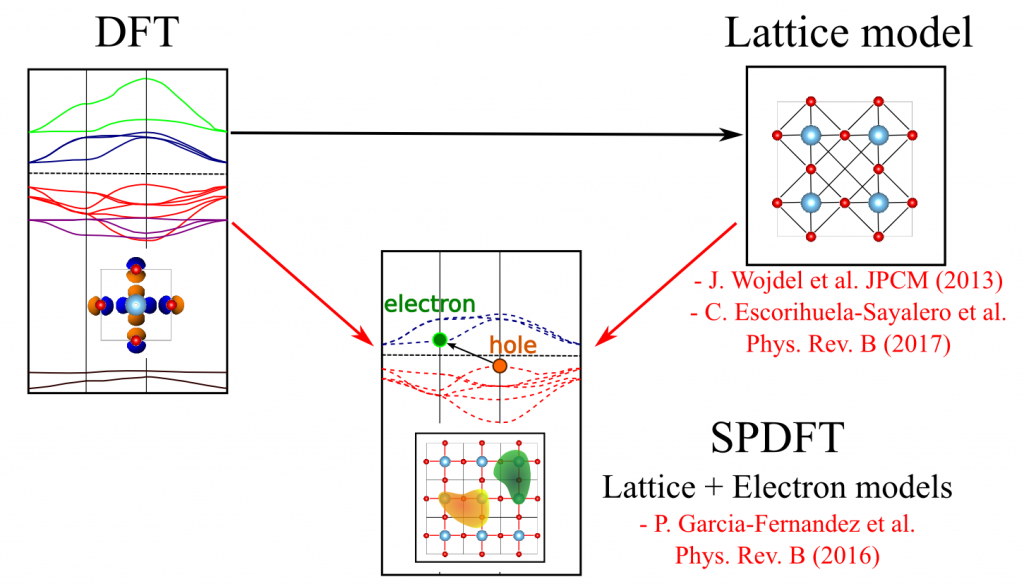
SPDFT allows approximating a DFT simulation including lattice and electron degrees of freedom with great precision!
Expanding the DFT energy in terms of the deformation density it is found that the zeroth-order term, corresponding to the DFT energy associated to the reference density, simply describes, in most cases, the energy surface of the unperturbed ground state. Higher order terms describe electron-hole excitations that allow obtaining the full range of electron properties like band, magnetic states, etc.
At difference with many other methods that are atom-based SPDFT is material -based. With this we mean that the basic construction block of a simulation is a model for a material. In particular, a SPDFT simulation requires the use of both a force-field and an electron Hamiltonian, formulated in a Wannier-function basis, that are obtained from first-principles simulations of a material in contrast with atom-atom potentials or electron basis based on atomic-functions typically used in other large-scale approaches. While this imposes the most important constraint to the method, the requirement of having a fixed bond topology – i.e. atoms cannot dissociate – this also allows the method to essentially match the accuracy of DFT even when dealing with very large number of atoms/electrons.
- Second-principles method for materials simulations including electron and lattice degrees of freedom, Pablo García-Fernández, Jacek C. Wojdeł, Jorge Íñiguez and Javier Junquera, Physical Review B 93, 195137 (2016). [arXiv:1511.07675]
Lattice potentials
In principles, SPDFT simulations can be based on any force field or effective potential treating all the atoms in the lattice. In SCALE-UP we implement a particular approach that adapts very well to our needs, as it can be trivially formulated for any material and permits a systematic improvement to reproduce a training set of first-principles data more and more accurately. This approach is based on the use of polynomial potentials that can be written as a Taylor series — in function of atomic distortions and lattice strains — around a convenient reference structure. The references describing this method, as implemented in SCALE-UP, are the following:
- First-principles model potentials for lattice-dynamical studies: general methodology and example of application to ferroic perovskite oxides, Jacek C. Wojdeł, Patrick Hermet, Mathias P. Ljungberg, Philippe Ghosez and Jorge Íñiguez, Journal of Physics: Condensed Matter 25, 305401 (2013). [arXiv:1301.5731]The basic formulation of the polynomial potentials was introduced in this 2013 article, with applications to ferroic perovskites PbTiO3 and SrTiO3.
-
Efficient systematic scheme to construct second-principles lattice-dynamical models, Carlos Escorihuela-Sayalero, Jacek C. Wojdeł and Jorge Íñiguez, Physical Review B 95, 094115 (2017). [arXiv:1608.06788]In this 2017 article, we implemented a new approach to the construction of polynomial models, taking advantage of one of its distinctive features — namely, that they are linear in the adjustable parameters — to introduce an ultra-fast fitting procedure that, in turn, allows us to select automatically the dominant interactions from a pool that includes essentially all impossible couplings. By doing this, we are able to routinely obtain very accurate models that reproduce a training set of DFT data within less than 1 meV per atom, and remain predictive (i.e., are cross-validated). This is the approach implemented in the utility to construct lattice models distributed with SCALE-UP.